Il bollettino del fegato n.8
n. 21 - Dipendenze: l'alcol resta il vero protagonista in Ticino n. 20 - Le malattie non virali del fegato n.19 - Dosaggio degli analgesici e cirrosi epatica n. 18 - Fegato e farmaci n. 17 - Il carcinoma epatocellulare (HCC) n. 16 - Fegato grasso n. 15 - Malattie epatiche e abuso di sostanze n. 14 - L'epatite B n.13 - L'epatite autoimmune n.12 SARS-CoV-2 (COVID-19) E FEGATO n.11 CEUS (Contrast Enhanced Ultrasound) n.10 Encefalopatia epatica n.9 - Epatite E n.8 - Morbo di Wilson n.7 - Biopsia epatica n.6 - Iperferritinemia nello studio medico n.5 - Epatopatia alcolica n.4 - Fegato grasso n.3 - Trattamento innovativo dell’ascite n.2 - La colangite biliare primitiva n.1 - I nuovi farmaci contro l'epatite CPRIMO PIANO
IL MORBO DI WILSON
La malattia di Wilson è dovuta a mutazioni del gene ATP7B sul cromosoma 13: esso codifica per una ATPasi di tipo P che risiede nell’apparato del Golgi degli epatociti. L'ATP7B è responsabile del trasporto del rame dalle proteine intracellulari alla via secretoria, composta da due direzioni: escrezione del rame nella bile o incorporazione del rame nell'apo-ceruloplasmina per la sintesi della ceruloplasmina funzionale.
Entrambe le funzioni di ATP7B risultano alterate nella malattia di Wilson; la conseguenza è l’accumulo del rame nei tessuti, in particolare nel fegato e nel cervello.
Si stima che nel mondo vi sia una prevalenza di 30 individui colpiti per milione di abitanti.
Più di 500 mutazioni sono state descritte nel gene responsabile della malattia, 380 delle quali sono state confermate avere un ruolo nella sua patogenesi.
LA PAROLA ALL'ESPERTO
Dr.ssa med. Antonella Robatto
Medico accreditato, Epatocentro Ticino
>> Visita il profilo
PRESENTAZIONE CLINICA
La malattia di Wilson può presentarsi a qualsiasi età, ma più frequentemente tra i 5 e i 35 anni.
La caratteristica clinica della malattia di Wilson è l'anello di Kayser-Fleischer, presente nel 95% dei pazienti con sintomi neurologici e in poco più della metà di quelli senza sintomi neurologici. Esso è causato dalla deposizione di rame nella membrana cornea di Desçemet. Per identificarlo è necessario un esame con lampada a fessura da parte di un oculista.
Nei bambini con malattia epatica, gli anelli di Kayser-Fleischer sono di solito assenti.
Gli anelli di Kayser-Fleischer non sono del tutto specifici della malattia di Wilson, poiché possono essere trovati in pazienti con malattie colestatiche croniche, compresi i bambini con colestasi neonatale.
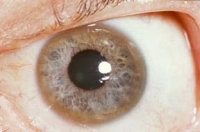
Anello di Kayser-Fleischer
Sintomi epatici
Malattie epatiche clinicamente evidenti possono precedere le manifestazioni neurologiche fino a 10 anni e la maggior parte dei pazienti con sintomi neurologici hanno un certo grado di malattia epatica all’esordio.
I sintomi della malattia epatica possono essere molto variabili; essi vanno dalla forma asintomatica, con solo anomalie biochimiche, alla cirrosi palese con tutte le sue complicanze.
La presentazione può essere indistinguibile da altre forme di epatite cronica attiva, con sintomi quali ittero, malessere e vaghi disturbi addominali.
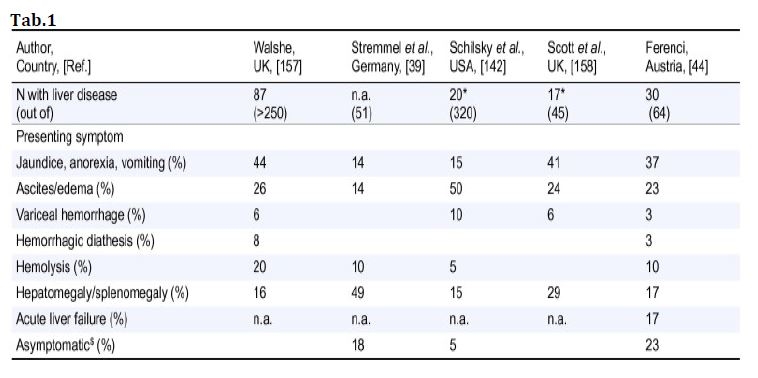
La malattia di Wilson può anche presentarsi come insufficienza epatica acuta a volte associata ad anemia emolitica Coombs-negativa e insufficienza renale acuta (vedi sintomi di insorgenza epatica tab.1).

Sintomi neurologici
La malattia di Wilson può manifestarsi con uno spettro impressionante di disturbi neurologici, comportamentali o psichiatrici: questi possono rappresentare la prima manifestazione clinica, e possono apparire contemporaneamente ai segni epatici, o qualche anno dopo.
La presentazione neurologica può essere blanda e intermittente per molti anni, ma può anche svilupparsi molto rapidamente, portando in pochi mesi alla completa disabilità. Le anomalie neurologiche possono essere classificate come segue:
1) Sindrome cinetico-rigido simile al morbo di Parkinson;
2) Pseudosclerosi dominata da tremore;
3) Atassia;
4) Sindrome distonica.
In molti casi, i segni neurologici sono molto difficili da classificare in quanto i pazienti possono avere più di un'anomalia, ognuna con diversi livelli di gravità.
Sintomi psichiatrici
I sintomi comportamentali e psichiatrici sono comuni e alcuni di essi possono precedere segni e sintomi neurologici o epatici. Circa un terzo dei pazienti presenta inizialmente anomalie psichiatriche.
Nei bambini con la malattia di Wilson si osservano un declino delle prestazioni scolastiche, cambiamenti di personalità, impulsività, umore labile, esibizionismo sessuale e comportamento inappropriato. I sintomi iniziali sono spesso mal diagnosticati come problemi comportamentali associati alla pubertà.
Nelle persone anziane si possono osservare caratteristiche psicotiche che assomigliano a paranoia, schizofrenia o depressione, ma sono comuni anche cambiamenti comportamentali. Un grave deterioramento cognitivo è osservato in pazienti con malattia neurologica avanzata, ma in generale, la funzione cognitiva non è notevolmente compromessa.
PROGNOSI
La malattia di Wilson non trattata è fatale.
Con il trattamento chelante e il trapianto di fegato, viene prolungata la sopravvivenza.
In generale, la prognosi per la sopravvivenza dipende dalla gravità della malattia epatica e neurologica e dalla compliance al trattamento farmacologico. La funzione epatica diventa normale in 1-2 anni di trattamento nella maggior parte dei pazienti senza cirrosi o cirrosi compensata all’esordio, e poi rimane stabile senza malattia epatica progressiva con adesione al trattamento.
Nei casi invece di pazienti con insufficienza epatica acuta, la terapia medica è raramente efficace a causa del tempo necessario per rimuovere il rame tossico dall'organismo.
Un indice prognostico è stato sviluppato e successivamente modificato da Dhawan et al. (vedi tab.1).
Un punteggio superiore a 11 è sempre fatale senza trapianto di fegato (Tab.2).


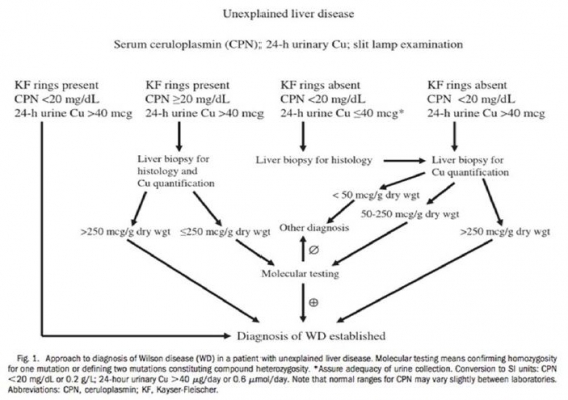
DIAGNOSI
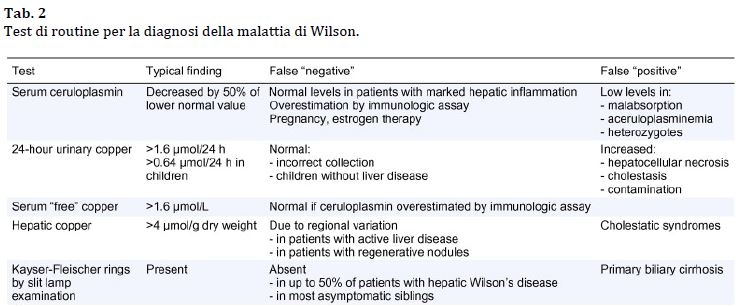
La combinazione dell’ anello di Kayser-Fleischer e un basso livello di ceruloplasmina sierica (<0.1 g/L) è di solito sufficiente per stabilire una diagnosi.
Quando gli anelli di Kayser-Fleischer non sono presenti, i livelli di ceruloplasmina da soli non sono sempre affidabili perché possono essere bassi per ragioni diverse dalla malattia di Wilson (es. epatite autoimmune, grave insufficienza epatica nella malattia epatica avanzata, celiachia, aceruloplasminemia familiare).
D'altra parte, l'infiammazione nel fegato o di altri organi può causare l'aumento della concentrazione di ceruloplasmina a livelli normali, riflettendo la sua identità di proteina in fase acuta
E’ spesso quindi necessaria una combinazione di test che confermino il metabolismo del rame disturbato (Tab.2).

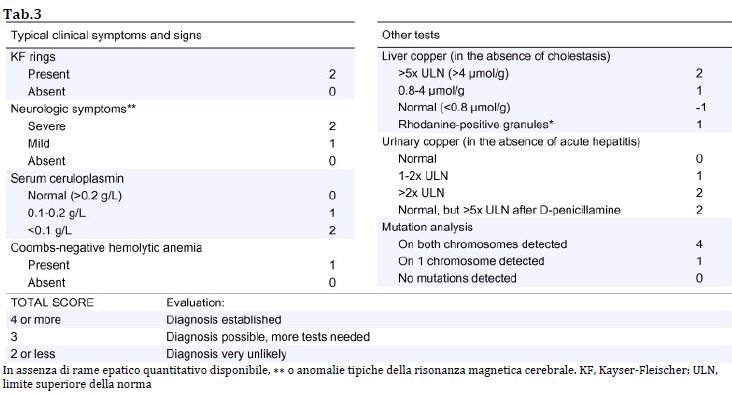
Anche lo score di Leipzig (2001) fornisce una buona accuratezza diagnostica (tab.3).

BIOPSIA EPATICA
A fini diagnostici, una biopsia epatica è necessaria solo se i segni clinici e gli esami non invasivi non consentono una diagnosi finale o se vi è il sospetto di altre o ulteriori patologie epatiche.
Le prime anomalie istologiche del fegato comprendono la steatosi lieve, i nuclei glicogeni negli epatociti e la necrosi epatocellulare focale, spesso, erroneamente diagnosticati come malattia epatica grassa non alcolica (NAFLD) o steatoepatite non alcolica (NASH). La biopsia epatica può mostrare le classiche caratteristiche istologiche dell'epatite autoimmune . Con il danno parenchimale progressivo si sviluppano la fibrosi e successivamente la cirrosi.
Per quanto riguarda l'individuazione del rame, soprattutto nelle prime fasi della malattia, esso non è sempre istochimicamente rilevabile. L'assenza di rame pertanto non esclude la malattia di Wilson.
Nell’analisi ultrastrutturale dei campioni di fegato l'alterazione più evidente è l'aumento dello spazio intracristale con dilatazione delle punte delle cristae, che crea un aspetto cistico.
In assenza di colestasi, questi cambiamenti sono considerati essenzialmente patognomonici della malattia di Wilson.
TEST GENETICI E SCREENING FAMILIARE
La diagnosi genetica molecolare diretta è difficile a causa di più di 500 possibili mutazioni. Tuttavia, è ragionevole eseguire l'analisi molecolare del gene ATP7B in qualsiasi paziente che abbia una diagnosi provvisoria della malattia di Wilson, sia a scopo di conferma che per facilitare il successivo screening dei familiari
È essenziale infatti controllare la famiglia dei pazienti che si presentano con la malattia di Wilson, perché la probabilità che un fratello sia omozigote - e quindi sviluppi una malattia clinica - è del 25%. Tra i figli, la probabilità è dello 0,5%. Sebbene questo rischio sia basso, l'analisi del gene ATP7B per le mutazioni nei figli di un paziente è giustificata dal potenziale decorso devastante della malattia di Wilson.
TRATTAMENTO
Per il trattamento della malattia di Wilson sono disponibili diversi farmaci, tra cui la D-penicillamina, la trientina, lo zinco, il tetrathiomolibdate e il dimercaprol. Una volta che la diagnosi è stata fatta, il trattamento deve durare tutta la vita.
TRAPIANTO DI FEGATO
Il trapianto è spesso necessario per pazienti con insufficienza epatica acuta o cirrosi scompensata. Poiché il difetto biochimico risiede principalmente nel fegato, il trapianto di fegato ortotopico (OLT) corregge il problema sottostante.
Un'osservazione limitata suggerisce che anche i sintomi neurologici dei pazienti che necessitano di OLT possono migliorare , anche se sono stati osservati casi di deterioramento neurologico nonostante un trapianto di successo.
GRAVIDANZA
Le donne con malattia di Wilson possono sostenere una gravidanza.
Lo stato di rame della paziente dovrebbe essere ottimizzato prima della gravidanza. Anche se c'è qualche preoccupazione sulla teratogenicità della D-penicillamina, i rischi di interrompere il trattamento superano quelli di continuarlo.
Questo vale anche per il trattamento con trientina o zinco.
L'allattamento al seno con terapia chelante non è raccomandato, anche se ci sono rapporti che indicano che i bambini allattati al seno da madri in terapia con D-penicillamina non hanno avuto problemi.
RACCOMANDAZIONI CONCLUSIVE
Qualsiasi malattia epatica di origine sconosciuta dovrebbe essere considerata come malattia di Wilson fino a prova contraria.
La malattia di Wilson entra nella diagnosi differenziale di ogni giovane paziente con epatite acuta.
Vista la gravità della patologia, una diagnosi tempestiva è fondamentale per la sopravvivenza e la qualità di vita dei pazienti.
ALGORITMO PER LA DIAGNOSI DI MALATTIA DI WILSON IN PAZIENTI CON EPATOPATIA INSPIEGATA (AASLD Practice Guidelines)

Per approfondimenti:
EASL Clinical Practice Guidelines (CPGs)
AASLD Practice Guidelines
GIHEP Resources for Gastroenterology & Hepatology
IN EVIDENZA
Anche senza danno epatico avanzato, la terapia per l'epatite C riduce il rischio di mortalità
Questi i risultati di un nuovo recente studio.
Scopri di più leggendo l'abstract dello studio e visitando il sito dell'American Association for the Study of Liver Disease (AASLD).
PUBBLICAZIONI
L’articolo scientifico “Autoimmune liver disease serology in acute hepatitis E virus infection”, redatto anche da medici dell’Epatocentro, è stato selezionato come “manoscritto del mese” nel Journal of Autoimmunity.
Lo studio è frutto del lavoro congiunto di diversi professionisti, tra i quali la Dr.ssa Benedetta Terziroli Beretta-Piccoli, il Prof. Dr. Andreas Cerny, la Dr.ssa Adriana Baserga e la Dr.ssa Claudia Di Bartolomeo, nonché della collaborazione di altri coautori di altri istituti ed enti ospedalieri.
L’articolo evidenzia come gli autoanticorpi siano frequentemente presenti durante le infezioni acute di virus epatico E (HEV), indicando come il virus epatico E dovrebbe essere escluso prima di diagnosticare l’epatite autoimmune (AIH).
FORMAZIONE
Gli incontri formativi proposti dalla Fondazione Epatocentro Ticino per il mese di dicembre.
Scarica il calendario